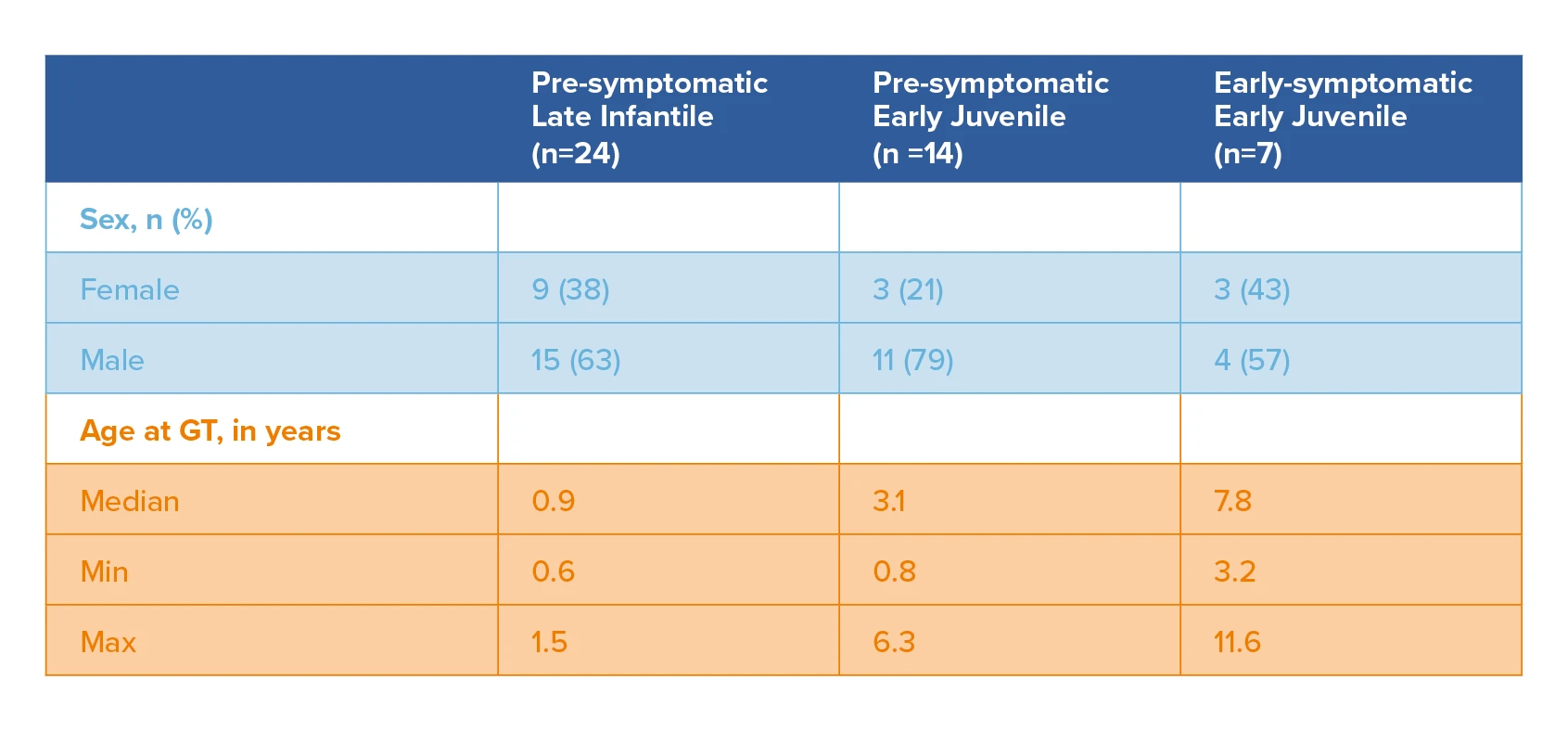
Clinical efficacy was evaluated in the integrated efficacy set (N=45), including pre-symptomatic late infantile (PSLI), pre-symptomatic early juvenile (PSEJ), early-symptomatic early juvenile (ESEJ) MLD patients treated with Libmeldy. This included patients treated in the Registrational Study (Study 201222), patients treated in a study with the commercial (cryopreserved) formulation (Study 205756), patients treated in the context of 3 expanded access programs, and patients enrolled in an observational long-term follow up study and treated either in the nominal compassionate use or the commercial setting. The median duration of post-treatment follow-up was 6.1 years in PSLI patients (range: 2.0 to 13.2 years), 3.3 years in PSEJ patients (range: 0.6 to 11.0 years) and 9.2 years in ESEJ patients (range: 0.5 to 10.9 years).1
Summary of demographic characteristics of the PSLI, PSEJ and ESEJ patients at time of gene therapy (Integrated efficacy set [N=45])
Libmeldy Summary of Product Characteristics.1
Primary endpoints
The co-primary endpoints were, at 2 years post-treatment:1-2
An improvement of 10% of the total GMFM-88 score in treated patients compared to natural history.
A significant (≥2 SD) increase of ARSA activity from pre-treatment values in PBMCs.
At year 3, an interim safety and efficacy analysis was planned and, at year 8, an efficacy and safety follow-up was planned.
Safety1,2
The safety of Libmeldy was evaluated in 49 patients with MLD. The median duration of follow-up was 5.6 years (range: 0.5 to 13.2 years). Three patients died and a total of 46 patients remained in the follow-up phase. All three deaths were not considered to be related to Libmeldy treatment.
Adverse reactions attributed to Libmeldy1
Most Adverse reactions were potentially related to busulfan conditioning or to MLD/disease progression
Anti-ARSA antibodies: All patients (N=39) in the clinical studies were tested for anti-ARSA antibodies (AAA) and 15% had a positive result. A similar percentage of patients treated either in the context of nominal compassionate use or commercial setting had positive AAA results reported. Antibody titres were generally low and the majority of cases resolved either spontaneously or after treatment with rituximab.
Risk of insertional oncogenesis: There is a theoretical risk of leukaemia or lymphoma after treatment with Libmeldy.
The long-term efficacy and safety of Libmeldy are unknown. Patients are asked to enrol in a follow up study for up to 15 years to better understand the long-term effects of Libmeldy.
Treatment with Libmeldy is preceded by medical interventions, namely haematopoietic stem cell collection through peripheral blood mobilisation with G-CSF with or without plerixafor followed by apheresis, and myeloablative conditioning (preferably using busulfan), which carry their own risks.
Libmeldy must be administered in a qualified treatment centre with experience in haematopoietic stem cell transplantation (HSCT).
Efficacy
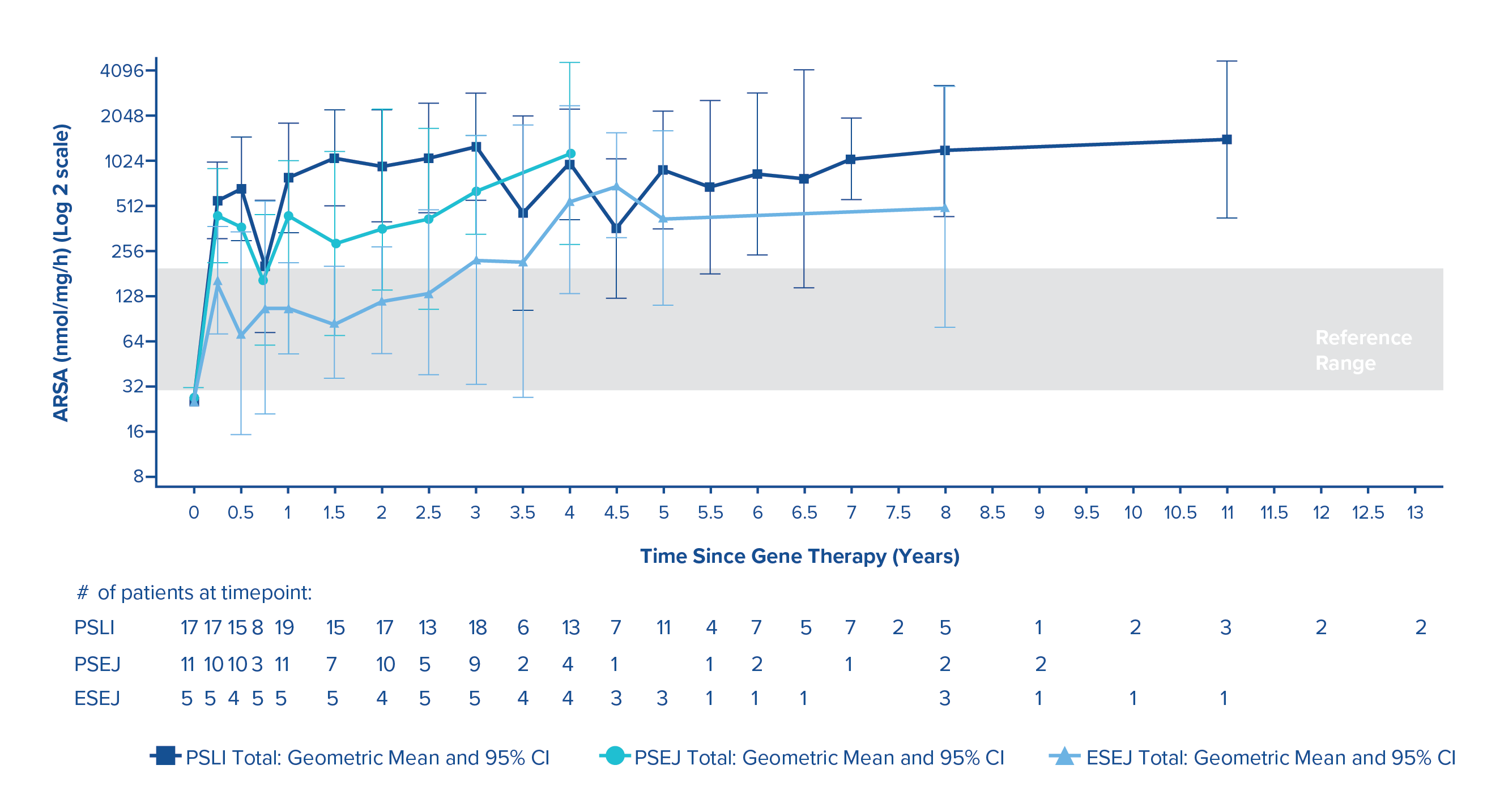
Normalised ARSA activity in peripheral blood mononuclear cells
Reconstitution of ARSA activity in the haematopoietic system was observed in all PSLI, PSEJ and ESEJ MLD patients treated in the clinical studies, with a progressive reconstitution of ARSA levels in PBMCs which reached geometric mean values within the normal reference range by 3 months post-treatment and remained stable within or above the normal range throughout the duration of the follow-up.1
ARSA activity in PBMCs over time (geometric mean and 95% CIs) in PSLI, PSEJ and ESEJ patients (N=35)1
Libmeldy Summary of Product Characteristics.1
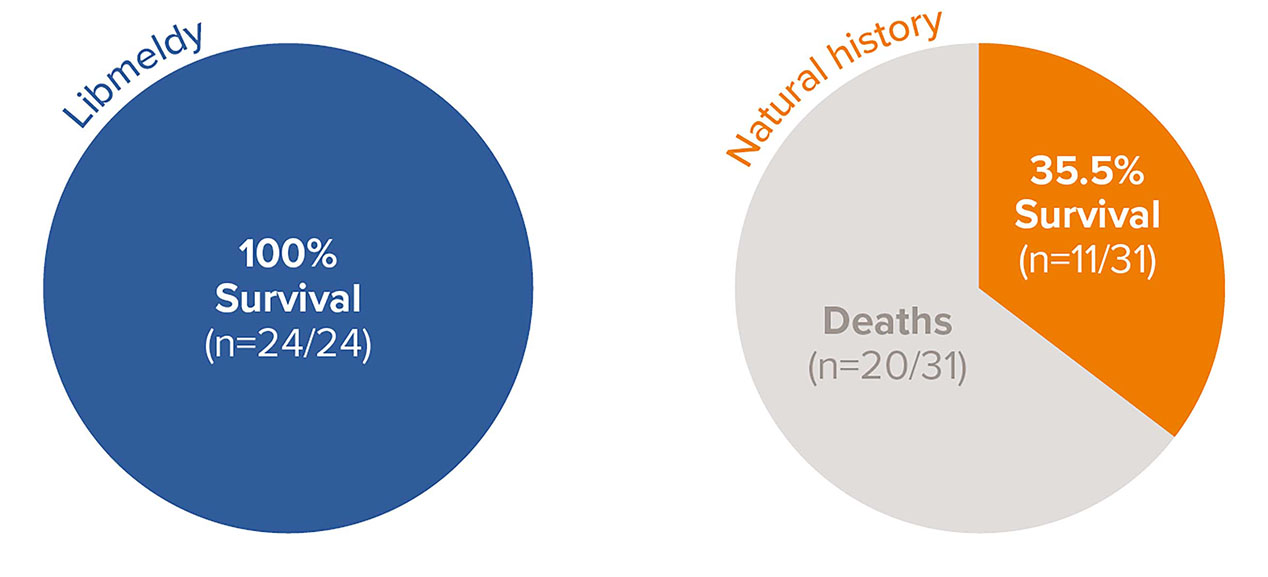
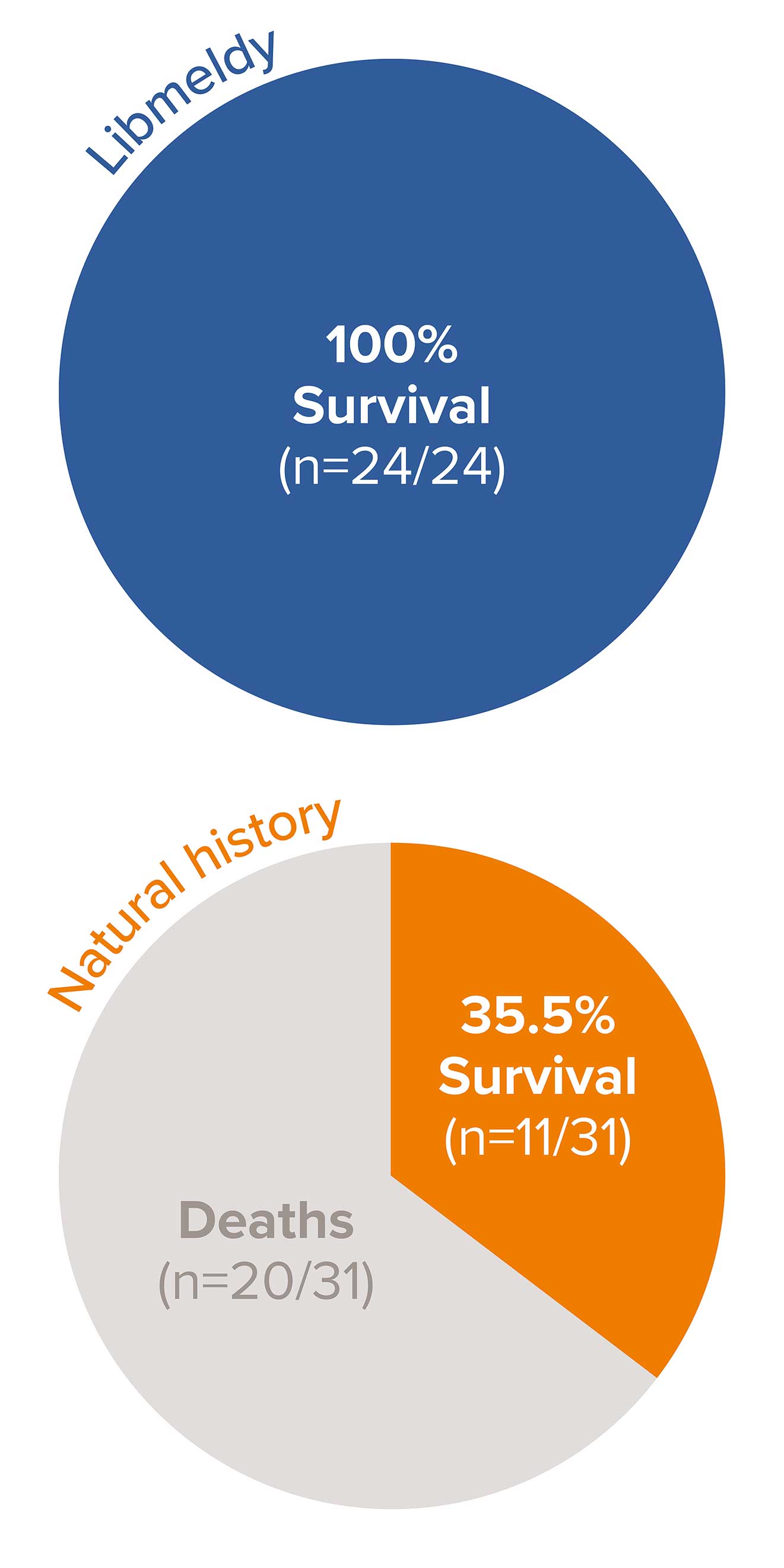
Survival benefit with Libmeldy vs. natural history group in pre-symptomatic late infantile (PSLI) MLD1
100% survival in 24 PSLI patients treated with Libmeldy, with the longest follow-up of 13.2 years post treatment vs. 35.5% survival (11/31) in untreated late infantile patients in the natural history cohort at the time of the analysis.1
Adapted from Libmeldy Summary of Product Characteristics.1
No treated PSLI patients have died, 20 of 31 NHx LI patients (65%) have died and the median actual age at death for the NHx LI patients was 5.9 years (range 3.5 to 13.4 years).
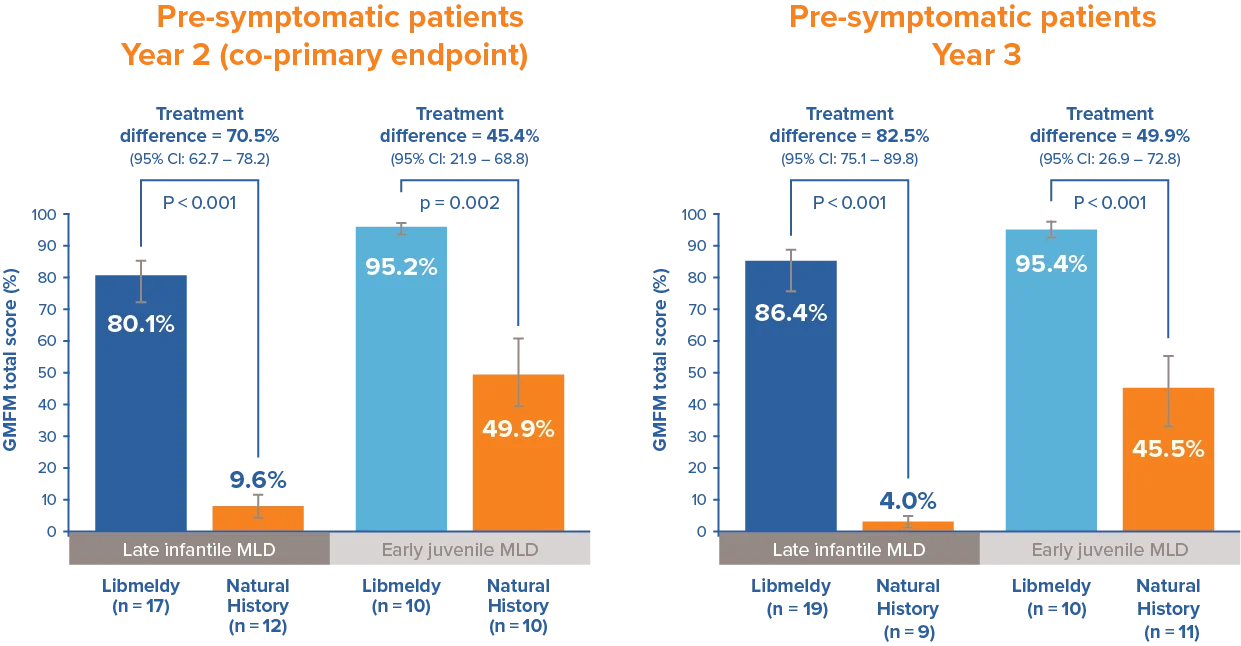
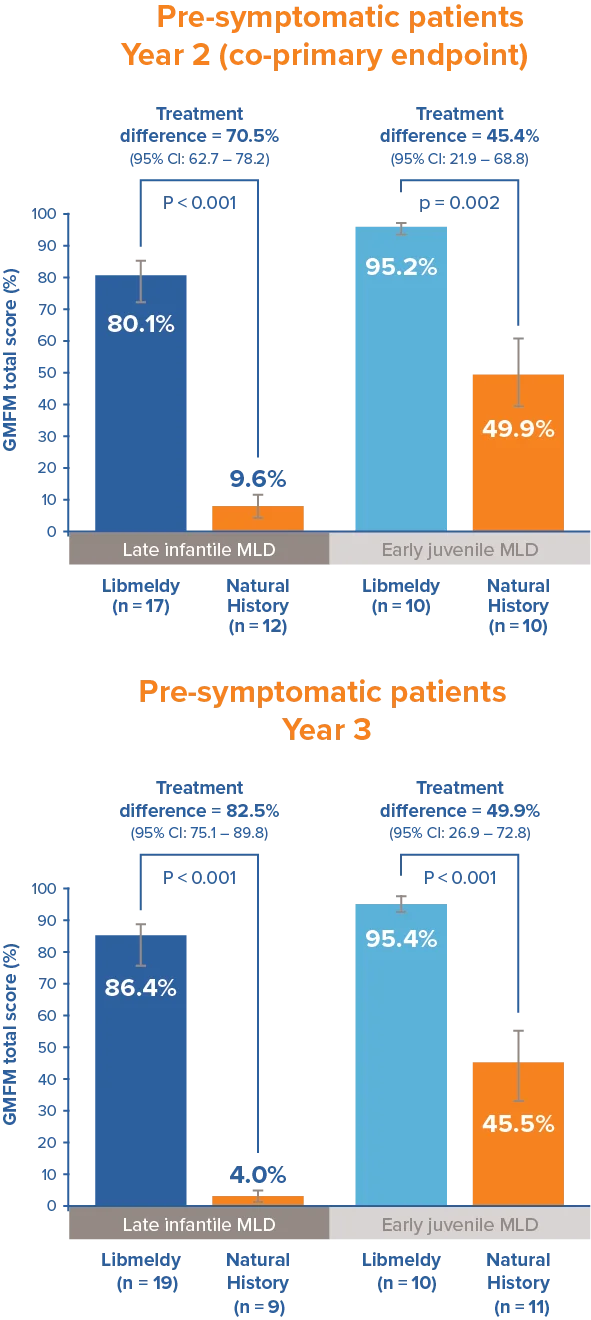
Statistically and clinically significant improvements in Gross Motor Function1
Patients treated with Libmeldy showed statistically and clinically significant improvements in Gross Motor Function measure at 24 months after treatment; when compared to natural history patients, results significantly exceed the pre-defined threshold of 10%.
Statistically significant differences were maintained at 3 years.1
Adapted from Libmeldy Summary of Product Characteristics.1
The Gross Motor Function Measure at 2 years after treatment was a co-primary endpoint of the registrational clinical study.
Note: Analysis of covariance adjusting for treatment and age. P-values are from a two-sided 5% hypothesis test with null hypothesis of 10% difference.
Gross Motor Function Measure (GMFM) is the most common functional outcome measure for gross motor functioning in children with neurologically based conditions.3 It is designed to measure gross motor function over time for children with disabilities, from 5 months to 16 years of age.2 The GMFM-88 test contains 88 questions grouped into five dimensions:3 lying and rolling (17), sitting (20), crawling and kneeling (14), standing (13), and walking, running and jumping (24). Percentage scores are calculated within each dimension and averaged to obtain a total score that ranges from 0 to 100. Higher scores indicate better capacity.3
Qualified treatment centres (QTCs) have the required infrastructure and experience in haematopoietic stem cell transplantation and the management of leukodystrophies to ensure the consistency and quality of treatment. For more details, see treatment process.
Abbreviations
CI: confidence interval; EJ: early juvenile; ES: early-symptomatic; GMFM: gross motor function measurement; GT: gene therapy; HSC: haematopoietic stem cell; LI: late infantile; MLD: metachromatic leukodystrophy; PBMC: peripheral blood mononuclear cells; PS: pre-symptomatic.
▼ This medicinal product is subject to additional monitoring. Healthcare professionals are asked to report any suspected adverse events. Further information about local reporting details can be found in Section 4.8 of the Summary of Product Characteristics.
Please also report any adverse events to Orchard Therapeutics at: drugsafety@orchard-tx.com
For any medical questions, please contact medinfo@orchard-tx.com
References
Libmeldy, Summary of Product Characteristics.
Fumagalli F, et al. Lancet 2022; 399(10322): 372-383.
Alotaibi M, et al. Disabil Rehabil 2014; 36(8): 617-627.