MLD is a rare, genetic, neurometabolic demyelinating lysosomal storage disorder caused by a deficiency of the arylsulfatase A (ARSA) enzyme due to mutations in the ARSA gene.1,2
MLD is inherited in an autosomal-recessive pattern – the overall carrier frequency being between 1:100 and 1:130.3

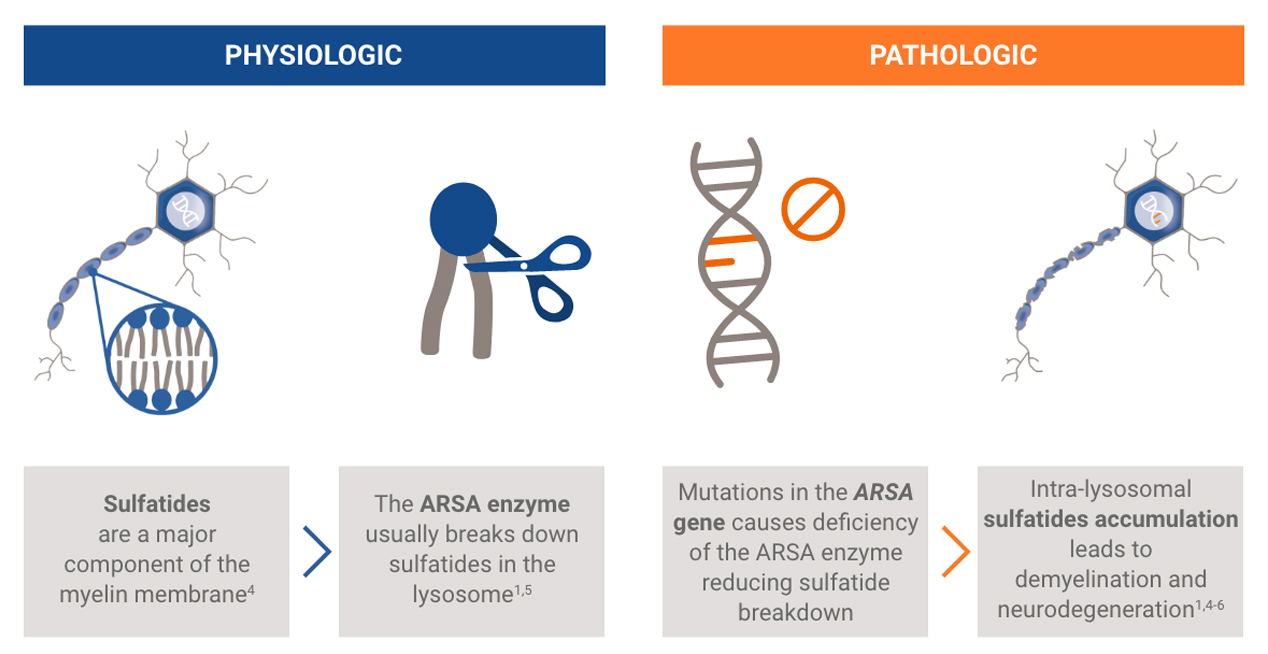
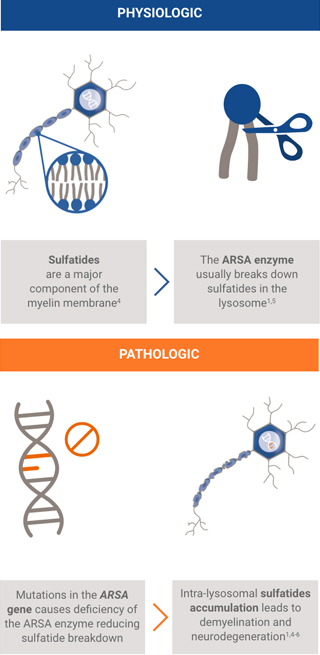

The ARSA gene encodes ARSA, a lysosomal enzyme necessary for the metabolism of sulfatides, a major component of the myelin membrane. Patients with MLD inherit two mutant alleles of the ARSA gene and have inadequate ARSA enzyme activity.
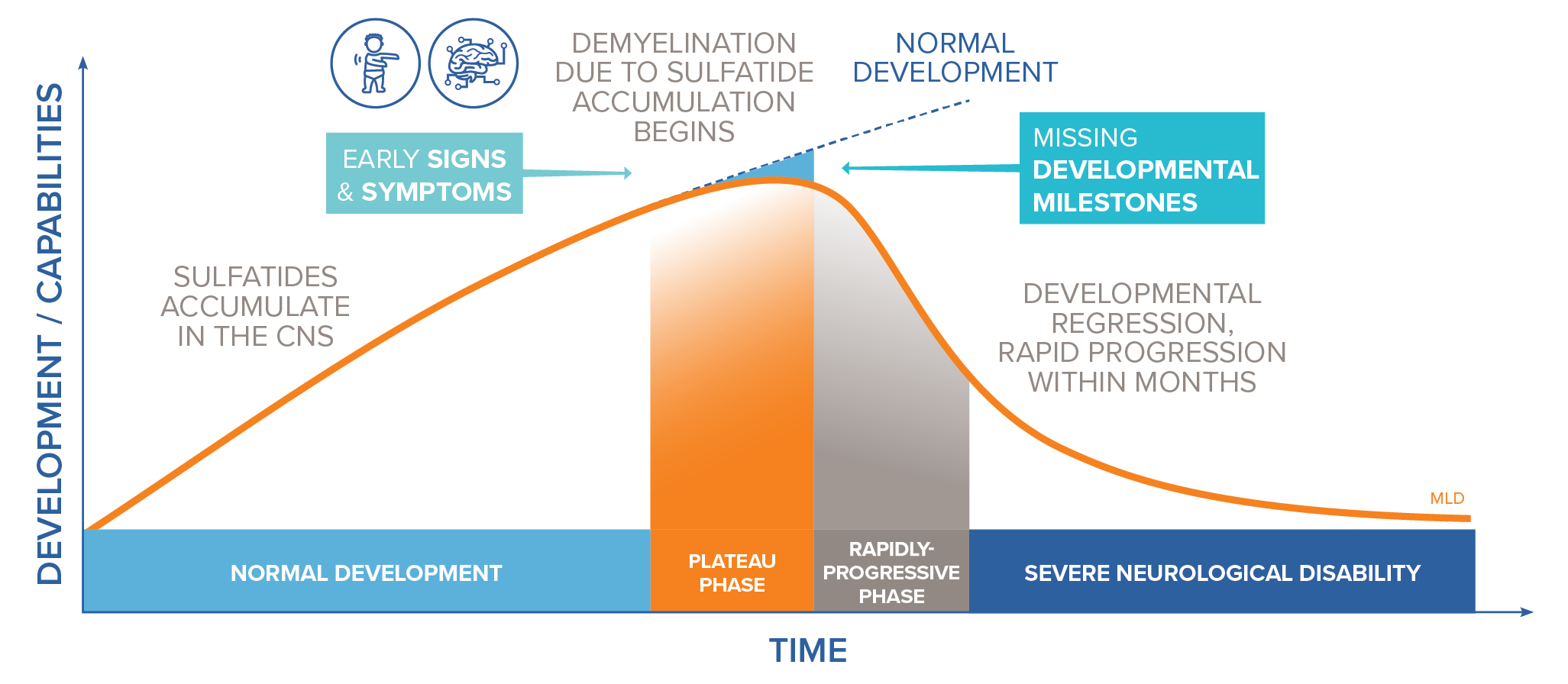
Damaging levels of sulfatides accumulate in lysosomes, leading to progressive demyelination that results in missed developmental milestones, regression, and ultimately leading to severe neurological symptoms.2 Patients experience severe motor and cognitive impairment, especially in the most common, early onset forms of the disease (late infantile and early juvenile forms).2
Adapted from von Figura K, et al., In: The metabolic and molecular bases of inherited disease, Vol. 3, 8th ed. New York: McGraw-Hill, 2001:3698-37247
Symptoms, age on onset and disease course vary, but patients progress to dysphagia, severe neurological disability and death.1,2
MLD has a considerable impact on the social, emotional and professional lives of patients and their families, including an average of 17 hours per day spent by families caring for their child with MLD.8
The global incidence of MLD is ~1.6 (range 0.6–2.5)/100,000 live births (for all forms of MLD).9
Recognising the early signs of MLD could present a challenge for several reasons:10
Early identification is critical. The progressive, irreversible nature of MLD demands an understanding of its clinical course and requires immediate, decisive action to prevent patient regression and improve overall outcomes.7
Early symptoms vary depending on whether the condition is late infantile (LI) or juvenile MLD.
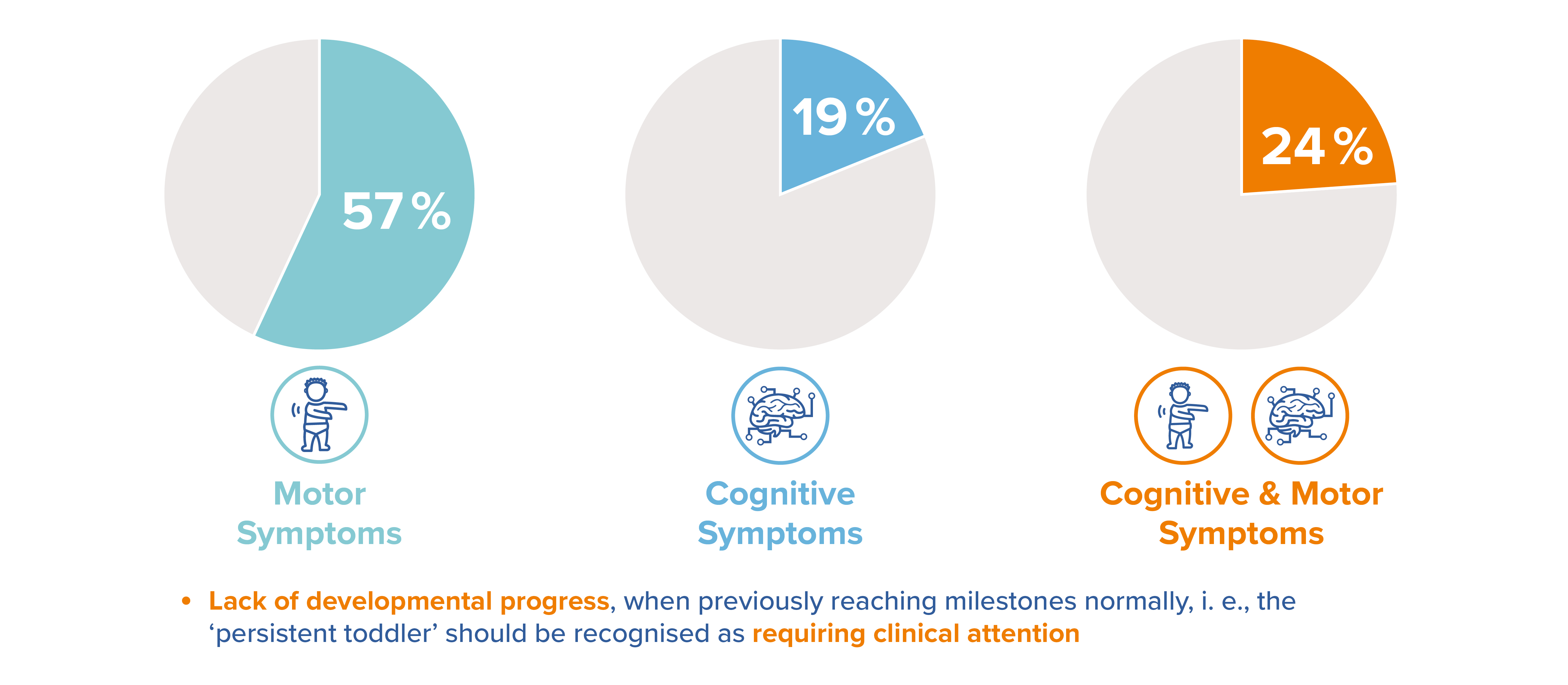

Adapted from Eichler et al. Initial Signs and Symptoms of MLD: A Caregiver Perspective. Poster presented at the Annual WORLD Symposium, February 8-12 202111
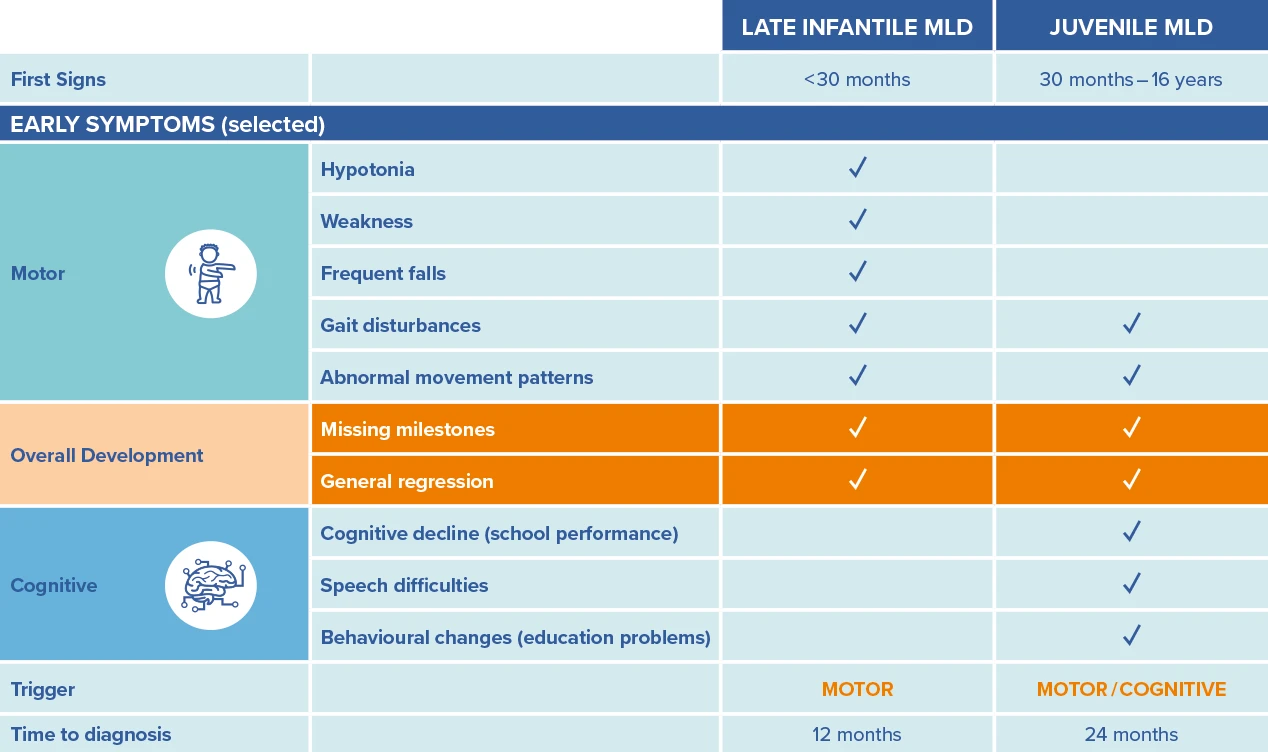
Adapted from von Figura K, et al., In: The metabolic and molecular bases of inherited disease, Vol. 3, 8th ed. New York: McGraw-Hill, 2001:3698-37247
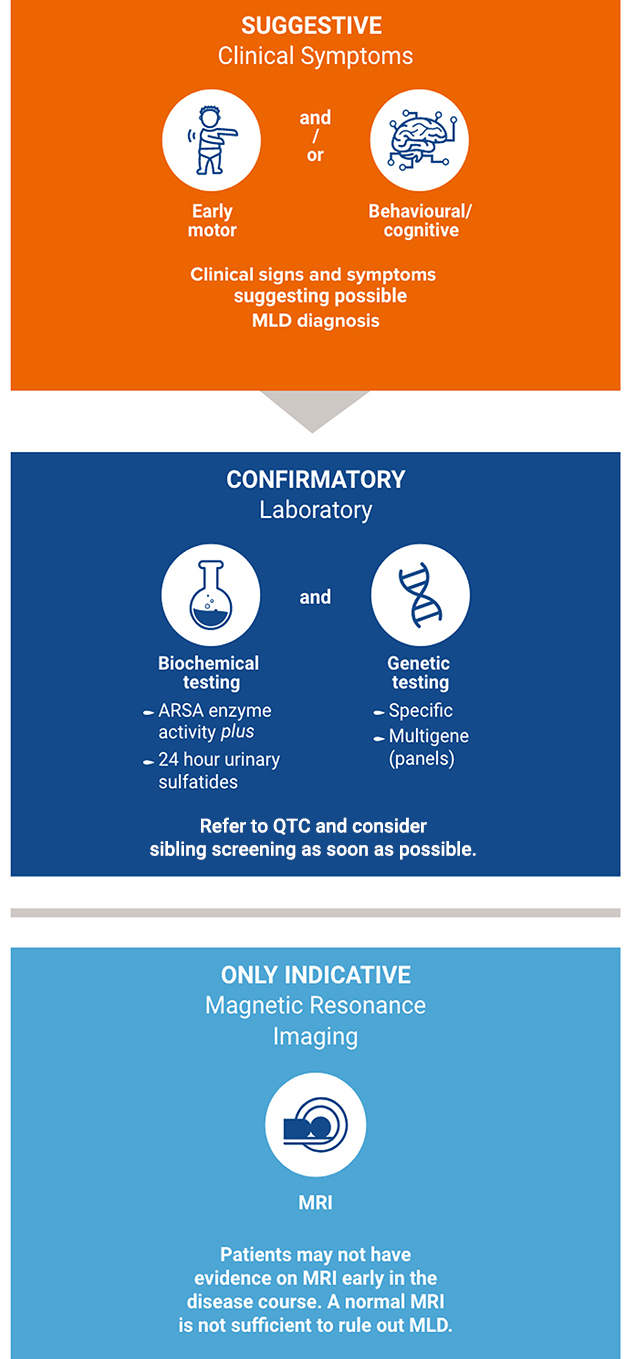
For HCPs with a symptomatic patient, the following process should be followed:12
The earlier patients can be identified, the better the outcomes. This can be achieved by family screening and newborn screening.12 Newborn screening compatible with established NBS infrastructures is available. Globally, implementation of MLD screening is gathering momentum and has been implemented as retrospective and prospective pilot studies as well as nationwide/statewide (USA) screening.19
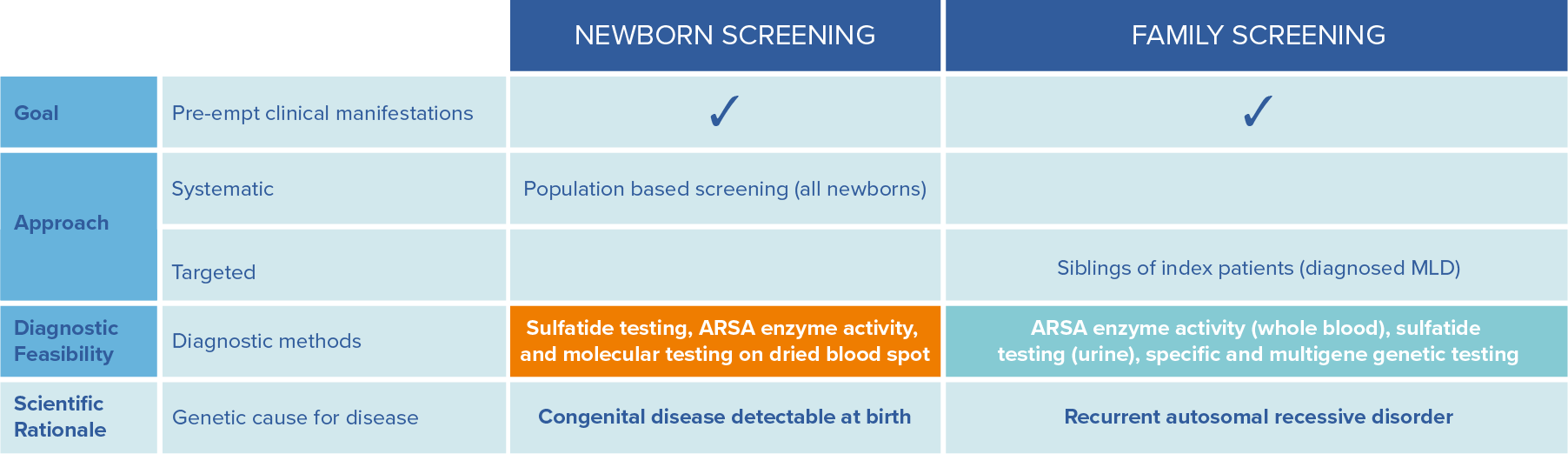
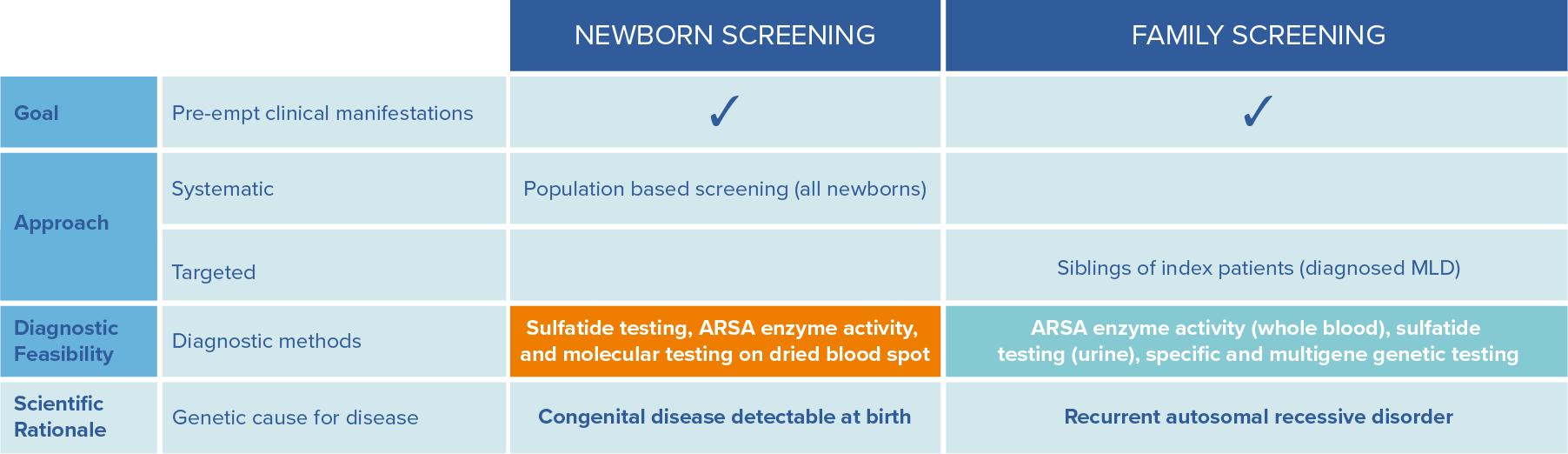
To allow a timely diagnosis and potential treatment, it is strongly recommended to initiate parallel family testing upon a strong suspicion of an MLD index case.12
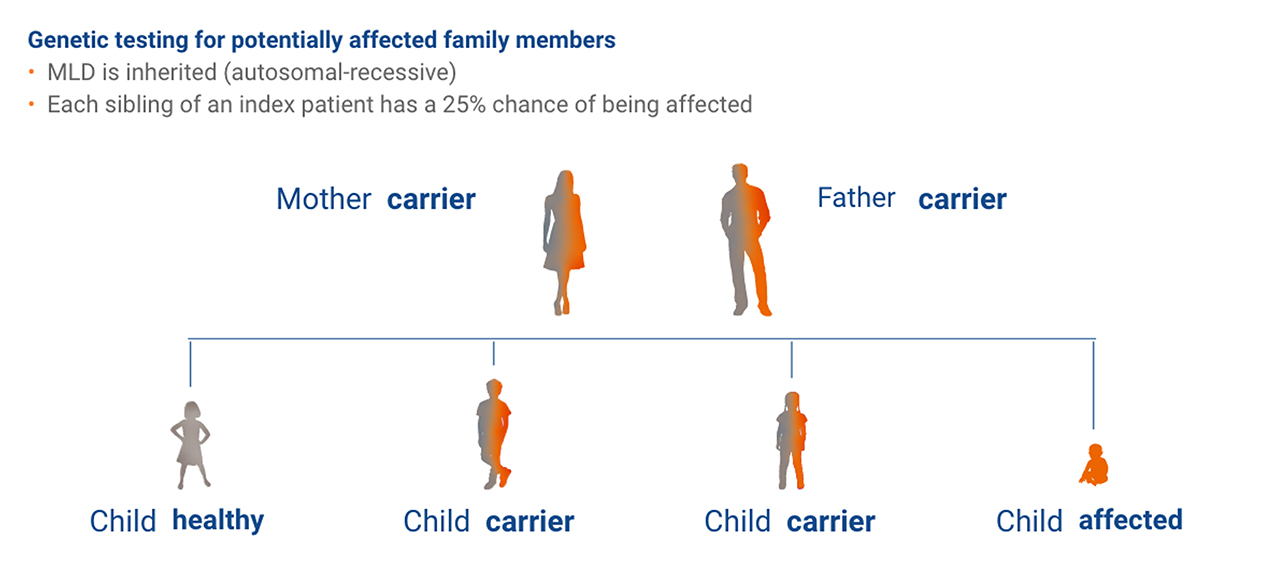
Adapted from Parikh et al., Molecular Genetics and Metabolism, 2015: 501-51512
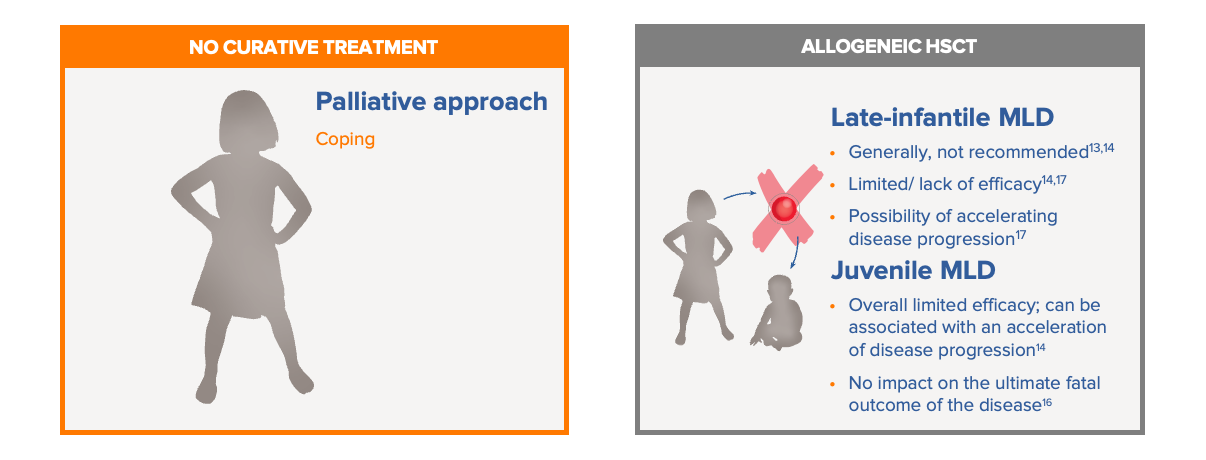
Before the approval of Libmeldy, there were limited treatment options for patients with MLD.13-16
Standard of care in MLD previously was limited to best supportive care, which involves the palliative treatment of symptoms. The use of allogeneic HSCT is only used in a minority of juvenile MLD patients. For many patients with MLD, particularly those with the late infantile, pre-symptomatic and early symptomatic form of the disease, allogenic HSCT is not recommended due to limited/no efficacy and inherent risks (including the possibility that pre-transplant procedures may even facilitate disease progression).17 Limitations of HSCT may include risk of graft-vs-host disease, limited levels of ARSA produced by the bone marrow HCST-derived microglia, limited donor match availability, and procedure-associated complications. Best supportive care and allogeneic HSCT are summarised below:13-16
Libmeldy is the first autologous haematopoietic stem cell (HSC) gene therapy product for early-onset MLD, as well as the first approved treatment.18
▼ This medicinal product is subject to additional monitoring. Healthcare professionals are asked to report any suspected adverse events. Further information about local reporting details can be found in Section 4.8 of the Summary of Product Characteristics.
Please also report any adverse events to Orchard Therapeutics at: drugsafety@orchard-tx.com
For any medical questions, please contact medinfo@orchard-tx.com
References
EM-002-2600005 | June 2026